Achievement
We proposed the numerical scheme and the algorithm to estimate its parameters which allow for the stable simulation of stiff multiscale chemical systems on time grids corresponding to the slowest scale of the system.
Significance and Impact
At the cellular level, even a single molecule can drastically impact the outcome of reactions. Markov chain models can account for both discrete and stochastic nature of such processes and provide convenient tools for the mathematical description of stochastic chemical kinetics. The evolution of probabilities in Markovian kinetic networks is entirely specified by the (chemical) master equation (CME) which can be solved only for very small or simple systems.
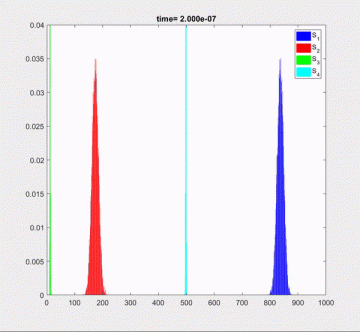
Simulation techniques provide an alternative to the master equation by generating large ensembles of possible temporal state paths. However, many biochemical systems contain both fast and slow reactions. It is known that transitory and highly reactive species involved in fast reactions may reach equilibrium and be asymptotically at a steady state within the coarse time scale of slow reactions. Sampling fast intermediate species from their quasi-stationary distributions using SSA is redundant on time intervals much exceeding the appropriate relaxation times. Tau-leaping methods applied to such systems usually require tiny steps in order to remain stable and are also inefficient. However, unlike deterministic dynamics, fast stochastic fluctuations represent an essential qualitative feature of biochemical systems and not an artifact of a numerical integrator. Efficient stiff solvers must be able to skip over the fast and stable reactions while capturing their stochastic influence on the slow species.
Research Details
-
proposed the new implicit split-step tau-leaping method which is both stable in the mean and can accurately resolve stationary distributions of the chemical species involved in fast reactions
-
proposed the algorithm for estimating parameters of the integrator admitting accurate simulation of the fast and stable reactions on the coarse time grids.
Overview
Tau-leaping is a family of algorithms for the approximate simulation of the discrete state continuous time Markov chains. A motivation for the development of such methods can be found, for instance, in the fields of chemical kinetics and systems biology. It is known that the dynamical behavior of biochemical systems is often intrinsically stiff representing a serious challenge for their numerical approximation. The naive extension of stiff deterministic solvers to stochastic integration often yields numerical solutions with either impractically large relaxation times or incorrectly resolved covariance. In this effort, we propose a splitting heuristic which helps to resolve some of these issues. The proposed integrator contains a number of unknown parameters which are estimated for each particular problem from the moment equations of the corresponding linearized system. We show that this method is able to reproduce the exact mean and variance of the linear scalar test equation and demonstrates a good accuracy for the arbitrarily stiff systems at least in the linear case.
Citation
Last Updated: January 15, 2021 - 12:43 pm